Depuis sa découverte en 1953 grâce aux données des diffractions aux rayons X obtenues par Rosalind Franklin, la structure à double hélice de l’ADN est incontestablement une des images les plus iconiques liées à la science et à la biologie. En conséquence, elle fait partie du patrimoine de connaissances communes de notre époque et elle permet d’expliquer bien des processus biologiques.
En effet, l’appariement en double hélice de l’ADN est crucial pour son rôle lié au stockage et à la réplication de l’information génétique. Cette structuration particulière est due au fait que les quatre briques élémentaires qui la composent (nommées nucléotides ou bases) peuvent s’apparier seulement de manière sélective : une thymine est associée à une adénine et une cytosine fait face à une guanine. En conséquence, la machinerie cellulaire pourra, à partir d’un brin d’ADN modèle, synthétiser sans erreur son brin complémentaire, assurant ainsi la réplication de l’ADN nécessaire à la prolifération des cellules.
De plus, cette succession de bases contient l’information génétique des cellules ou de l’organisme, qui amènera à la production de protéines spécifiques. Par ailleurs, l’organisation des bases de l’ADN permet aussi de délimiter des régions codantes (c’est-à-dire les régions traduites en protéines, aussi appelées gènes) et des régions non codantes.
Néanmoins, la traduction de l’information de l’ADN en protéines ne peut pas se faire directement. Elle nécessite de passer par une structure intermédiaire : l’ARN messager (ARNm), très proche de l’ADN moyennant quelque modifications chimiques dans son squelette et la substitution de la thymine par l’uracile. Lues trois par trois (on parle de triplets), les bases constituant l’ARN messager correspondent à des acides aminés précis, qui sont les éléments de base des protéines. C’est ce code génétique qui permet la synthèse de l’ensemble des protéines.
Si l’ADN est pensé pour le stockage à long terme de l’information, l’ARN messager quant à lui est très labile et rapidement détruit par les cellules. Un de ses rôles majeurs est de servir de messager pour permettre la production des protéines. Pour ce faire, il n’est pas structuré en double brin (comme l’ADN), mais en simple brin. Cela lui confère une flexibilité particulière et la possibilité d’assumer différentes structures locales, comme de courtes doubles hélices ou des épingles à cheveux. Par ailleurs, à côté de l’ARN messager, on peut dénombrer d’autres types d’ARN exerçant des fonctions de contrôle ou de régulation, comme les ARN de transfert (ARNt) ou les ARN ribosomique (ARNr).
L’ADN, l’ARN et les protéines sont donc à la base de la biologie moléculaire moderne. Cependant, les mécanismes cellulaires qui entrent en jeu dans la réplication et l’expression de l’information génétique (la lecture des gènes qui aboutit à la formation des protéines) sont infiniment plus complexes. Les scientifiques se mobilisent depuis de nombreuses années afin de répondre aux interrogations suivantes : comment l’ADN s’organise-t-il dans nos cellules ? Quels phénomènes régulent l’expression des gènes ? Quel intérêt y a-t-il à contrôler l’expression des gènes ?
Comment l’ADN s’organise-t-il dans nos cellules ?
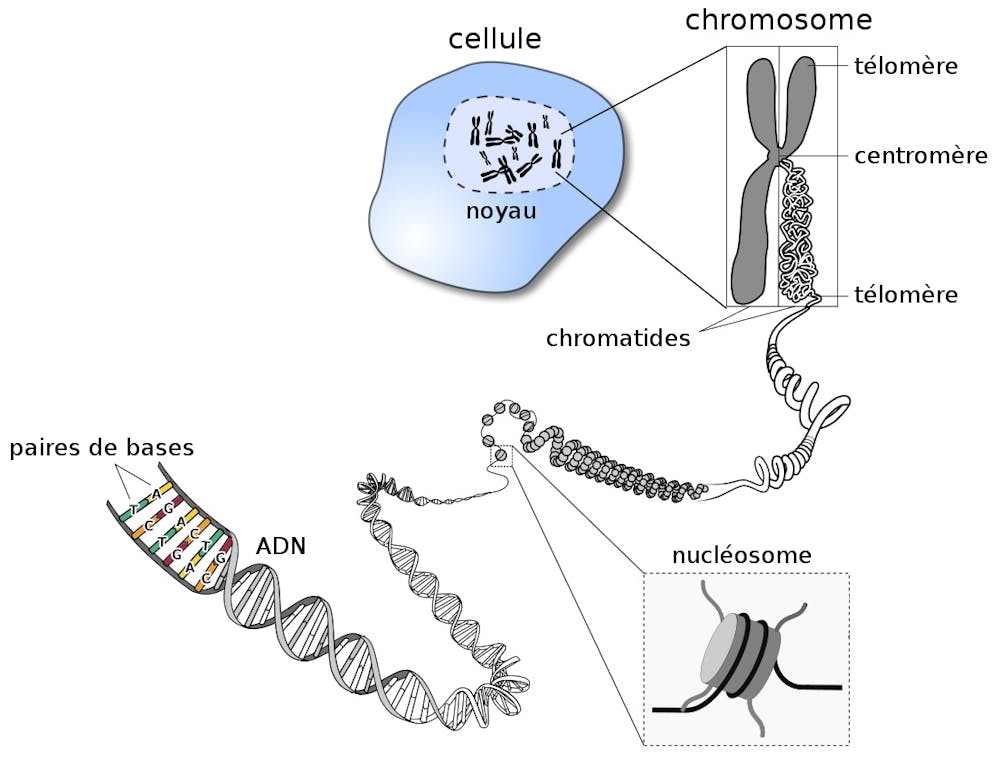
Dans le cas d’Homo sapiens, c’est-à-dire l’espèce humaine, l’ADN compte à peu près 3 milliards de bases. Plus simplement, si on mettait bout à bout l’ADN d’une seule de nos cellules, il serait long d’environ deux mètres, la taille standard d’un joueur de basket. Or, l’ADN se trouve exclusivement dans le noyau de chacune de nos cellules, qui mesure quelques micromètres (mille fois plus petit qu’un millimètre). Mais alors, comment deux mètres d’ADN peuvent-ils être contenus dans un millième de millimètre ?
La clé de ce mystère se situe dans la capacité de l’ADN à être très fortement compacté grâce à des protéines appelées histones. Les histones et l’ADN ont des affinités mutuelles très fortes grâce aux charges positives des premières, qui attirent les charges négatives du second. Dans nos cellules, l’ADN peut donc s’enrouler autour de groupes d’histones pour former un premier niveau de compaction : le nucléosome. Ce dernier est constitué d’environ 145 bases enroulées sur un cœur de quatre types d’histones (H2A, H2B, H3 et H4). La compaction de l’ADN autour des histones permet non seulement de faire rentrer le génome dans le noyau de la cellule, mais confère également une protection contre les sources externes de stress, comme les rayons UVs, les radicaux libres ou les radiations.
Par ailleurs, on retrouve de telles protéines de compaction de l’ADN dans des organismes plus simples (comme les bactéries). Elles sont alors appelées par le terme anglais de « histones-like ». Bien que plus petites que les histones, elles permettent elles aussi d’exercer sur l’ADN une force très importante pour maintenir sa compaction et protéger son intégrité.
Mais le nucléosome n’est que la première étape de compaction pour arriver à loger l’ADN d’une cellule dans son noyau. Les nucléosomes successifs vont ensuite s’organiser entre eux, de manière plus ou moins compacte, pour donner lieu au deuxième niveau d’organisation de l’ADN : la chromatine. Cette dernière va ensuite, à son tour, s’organiser dans des structures de taille encore plus larges que sont les chromosomes.
{kind=link}
{kind=link}
La structuration de l’ADN autour des histones n’est pas figée : elle possède au contraire un comportement dynamique très fort qui permet au génome d’être accessible et lu quand besoin est.
Pour pouvoir étudier ces phénomènes dynamiques, les scientifiques ont développé tout un panel de techniques : la cristallographie aux rayons X, la résonance magnétique nucléaire, ou encore la cryo-microscopie électronique. De même, l’utilisation des méthodes de simulation informatique ou mathématique permet, tel un microscope ultime, de visualiser, grâce à l’utilisation de supercalculateurs massivement parallèles, l’agencement des atomes dans l’espace et son évolution au cours du temps.
Quels phénomènes régulent l’expression des gènes ?
La compaction des nucléosomes en chromatine est donc hautement dynamique, et deux états distincts ont été identifiés : l’hétérochromatine, très compacte, et l’euchromatine, plus éparse. Sous forme d’hétérochromatine, le génome est moins accessible et il est impossible aux cellules de traiter ces parties de l’ADN pour produire l’ARN messager et donc les protéines. Ces gènes ne seront donc pas, ou très peu, exprimés. En revanche, l’euchromatine permet aisément l’expression des gènes et donc la production des protéines.
La dynamique de la chromatine ayant un rôle majeur sur le contrôle de l’expression des gènes, sa régulation précise est essentielle pour permettre la survie des cellules. Ceci est d’autant plus marqué chez les organismes pluricellulaires (constitués de plusieurs cellules), chez qui la production spécifique de protéines permet aux cellules de se différencier en cellules distinctes (nerveuses, musculaires, cutanées…) qui portent toutes le même matériel génétique, mais possèdent des propriétés très différentes.
Cependant, le contrôle de l’expression des gènes, et donc des protéines produites, n’est pas figé dans le temps. Au contraire, les cellules doivent pouvoir modifier le niveau de production des protéines en fonction de leur état (prolifération ou différenciation par exemple), mais aussi en réponse à des sources de stress. Par exemple, suite à une exposition incontrôlée aux radiations UV, qui endommagent la structure de l’ADN, les cellules devront augmenter la production des enzymes de réparation des lésions pour éviter leur accumulation. La transition réversible d’hétérochromatine à euchromatine (ou vice-versa) permet donc aux cellules de réagir aux modifications internes ou environnementales.
Mais alors, comment les cellules peuvent réguler la transition d’un état à l’autre ?
Pour répondre à cette question fondamentale, il est important de se rappeler que, contrairement aux idées reçues, seulement 3 % de l’ADN humain code activement pour des protéines. Le reste est dormant ou utilisé comme régulateur de l’expression des gènes. Des modifications chimiques de la structure des régions non codantes de l’ADN peuvent donc être utilisées pour réguler le niveau de la compaction de la chromatine, et donc l’expression des gènes. L’ensemble de ces modifications constitue les signaux épigénétiques, c’est-à-dire des modifications réversibles de l’ADN régulant l’expression des gènes.
Dans ce contexte, l’ajout d’un groupement méthyle sur la cytosine, qui donne lieu à la 5-méthylcytosine, représente un des signaux les plus importants. En effet, l’accumulation de 5-méthylcytosines dans des régions spécifiques riches en cytosine et guanine, appelées îlots CpG, va constituer un signal de compaction de la chromatine autour d’un gène particulier et donc en empêcher son expression. Pour moduler les niveaux de 5-méthylcytosine, les cellules mettent en œuvre des phénomènes chimiques complexes et très finement régulés, qui permettent d’introduire et d’éliminer réversiblement le groupement méthyle en passant par différentes étapes intermédiaires.
Par ailleurs, d’autres signaux épigénétiques existent, notamment les modifications chimiques des histones, qui participent également à moduler le niveau de compaction de la chromatine.
Contrôler l’expression des gènes : vers des thérapies épigénétiques
Les mécanismes moléculaires responsables du contrôle épigénétique doivent être parfaitement huilés pour permettre la survie des cellules, et leur dérégulation entraîne le développement de pathologies graves. Par exemple, une majorité des cancers sont liés à des profils épigénétiques aberrants, résultant de la diminution de l’expression de gènes qui empêchent le développement tumoral (gènes suppresseurs de tumeurs) et à l’augmentation des gènes qui, au contraire, le favorise (les oncogènes). Ces derniers confèrent, par exemple, la capacité de prolifération incontrôlée ou d’invasivité aux cellules cancéreuses.
Des stratégies thérapeutiques anticancéreuses basées sur la régulation épigénétique commencent donc à être développées pour cibler l’expression aberrante du profil génétique. Les thérapies épigénétiques actuellement en phase d’étude clinique se basent sur l’utilisation des médicaments capables de bloquer les enzymes responsables de l’élimination du groupement méthyle de la 5-méthylcytosine, pour potentiellement réactiver l’expression de gènes suppresseurs de tumeurs. Cependant, de par leur nature, ces enzymes ne sont pas spécifiques vis-à-vis des séquences de l’ADN, elles ne peuvent donc pas cibler de façon précise un gène particulier dans le génome entier. Ce manque de sélectivité se traduit par des effets secondaires très lourds des médicaments épigénétiques, ce qui limite fortement leur utilisation.
Alors, comment arriver à développer des stratégies permettant une plus grande sélectivité ? L’idée de base est de changer de cible : au lieu de viser les enzymes, il s’agirait de déméthyler directement les parties de l’ADN (permettant leur réexpression), grâce à des molécules capables d’induire la réaction chimique d’élimination du groupement méthyle.
Dans ce contexte, nous avons récemment déposé un brevet faisant état des premiers agents déméthylant directs de l’ADN, tout en montrant une efficacité contre la prolifération de différents types de cellules cancéreuses. Des études in-vivo et toxicologiques sont actuellement en cours en vue d’une future étape clinique. Cependant, pour cibler des gènes spécifiques, il est nécessaire d’adjoindre à ces molécules des groupements capables de reconnaître de manière précise des segments d’ADN. Pour atteindre cet objectif, il est nécessaire d’utiliser des approches combinées qui font intervenir, à côté des expériences de chimie et biologie moléculaire, la modélisation et la simulation moléculaire. En permettant de visualiser à l’échelle des atomes les interactions mises en jeu, la simulation moléculaire permettra de designer des molécules capables de cibler spécifiquement les régions d’intérêt, et donc diminuer les effets secondaires de ces médicaments.
Comme souvent en biologie, un phénomène qui paraît simple, en ce cas la structure et la fonction de l’ADN, cache en réalité de multiples facettes qui sont souvent méconnues mais néanmoins fondamentales. Ces aspects complexes permettent de comprendre les régulations très fines liées au fonctionnement des systèmes cellulaires, comme la régulation de l’expression des gènes. La simulation moléculaire est aujourd’hui un acteur clé de cette histoire, et elle le sera d’autant plus dans le futur. Les méthodes de biologie in-silico complémentent efficacement les techniques plus traditionnelles in-vitro et in-vivo, permettent de répondre à des questions biologiques clés, et se révèlent cruciale dans l’effort pour développer des médicaments plus performants. La biologie computationnelle est donc toute disposée à un resplendissant avenir dans la recherche scientifique et médicale.
Antonio Monari, Professeur en Chimie Théorique, Université de Paris; Emmanuelle Bignon, Chercheuse en biochimie computationnelle, Université de Lorraine et Stéphanie Grandemange, Professeur en biologie cellulaire, Université de Lorraine
Cet article est republié à partir de The Conversation sous licence Creative Commons. Lire l’article original.